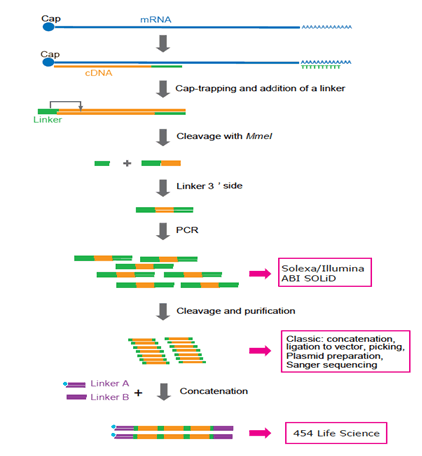
(B) Graph shows the comparison of the contig length of three assemblies plotted against the N statistic of the assembly [for instance, N40 (x-axis) is equal to about 1 Kbp (y-axis), which means that (10040=60) % of the entire assembly is contained in contigs no shorter than 1 Kbp]. RCC307 (Cyanobacteria), and Synechoccocus sp. We evaluated the type and frequency of errors in assembled contigs from metagenomic data using both a comparative and a reference genome approach. Shared reads were defined as those that mapped on reads of the other dataset using Bowtie with default settings [25]. Next generation sequencing (NGS) technologies, such as the Roche 454, Illumina/Solexa, and, to a lesser extent, ABI SOLiD, have been cornerstones in this revolution [5], [6], [7]. The percent of the reference genome recovered by these fragments as a fraction of the total length of the reference assembly was calculated using a custom Perl script. For more information about PLOS Subject Areas, click Velvet was used to assemble each of these Illumina datasets with K-mer set at 31. The average G+C% content of the metagenome was 47.4%; thus, our results are not simply attributable to higher abundance of A's and T's in the metagenome. Discover a faster, simpler path to publishing in a high-quality journal. Our previous evaluation showed that our hybrid protocol outperforms other approaches for assembling metagenomic and genomic data [18]. https://doi.org/10.1371/journal.pone.0030087.g001. Panels A and C represent the variation observed in reads from different (replicate) datasets of the same genome; red bars represent the median, the upper and lower box boundaries represent the upper and lower quartiles, and the upper and lower whiskers represent the largest and smallest observations. 7). Hence, the majority of non-homopolymer-associated errors remain challenging to model and thus, to correct.
 The frequency of single-base errors decreased with higher coverage of the corresponding contigs, i.e., the frequency dropped by about ten fold in contigs with 20 coverage relative to contigs with 2 coverage, reaching a plateau at about 20 coverage. Nine Illumina and eight Roche 454 assemblies from independent replicate datasets of the Fibrobacter succinogenes subsp. We compared the reads from the Lanier.Illumina dataset against the Lanier.454 dataset to identify the fraction of reads shared between the two datasets. For instance, searching all genes shared between the two assemblies against NCBI's Non Redundant (NR) protein database (Blastx) returned more complete matches with the Lanier.Illumina than the Lanier.454 data, regardless of the identity and e-value threshold used (14% more on average; Fig. The higher sequence error rate observed for the TIGR reference genome might be due to the different strain of F. succinogenes sequenced or differences in the sequencing platforms or the assembly protocols used by JGI and TIGR.
The frequency of single-base errors decreased with higher coverage of the corresponding contigs, i.e., the frequency dropped by about ten fold in contigs with 20 coverage relative to contigs with 2 coverage, reaching a plateau at about 20 coverage. Nine Illumina and eight Roche 454 assemblies from independent replicate datasets of the Fibrobacter succinogenes subsp. We compared the reads from the Lanier.Illumina dataset against the Lanier.454 dataset to identify the fraction of reads shared between the two datasets. For instance, searching all genes shared between the two assemblies against NCBI's Non Redundant (NR) protein database (Blastx) returned more complete matches with the Lanier.Illumina than the Lanier.454 data, regardless of the identity and e-value threshold used (14% more on average; Fig. The higher sequence error rate observed for the TIGR reference genome might be due to the different strain of F. succinogenes sequenced or differences in the sequencing platforms or the assembly protocols used by JGI and TIGR.
To provide new insights into these issues, we evaluated the two most frequently used platforms for microbial community metagenomic analysis, the Roche 454 FLX Titanium and the Illumina GA II, by comparing and contrasting reads and assemblies obtained from the same community DNA sample. The two platforms agreed on over 90% of the assembled contigs and 89% of the unassembled reads as well as on the estimated gene and genome abundance in the sample (Fig. 1B).  Assemblies of isolate genome sequences (closed or high-draft) were downloaded from the NCBI RefSeq database (called reference assemblies for convenience); raw Illumina and Roche 454 sequencing reads were available through the Joint Genome Institute (JGI, www.jgi.doe.gov). No, Is the Subject Area "Gene sequencing" applicable to this article? 2). Conceived and designed the experiments: CL NK KTK. 3), low G+C% genomes sequenced with this platform may have 20% or more genes with frameshift errors whereas the Illumina platform is not affected as much by the G+C% of the sequenced DNA (Fig. It is possible that the remaining 10% of the contig sequences might have been different because of imperfect or uneven splitting of the original DNA sample into the two aliquots sequenced and the fact that the diversity in the sample was not saturated by sequencing (estimates based on rarefaction curves using raw reads indicated that we sampled about 8085% of the total diversity in the Illumina data). Thus, Roche 454 is advantageous with respect to gene calling when working with unassembled reads. Consistent with the metagenomic observations, we found that Roche 454 assemblies from genome data contained a significantly higher portion of frameshift errors compared to Illumina assemblies from the same genome, when the assemblies were built with 5 times more Illumina data than the Roche 454 data, matching the relative ratio of the metagenomic data reported above. We identified 0.4 million homopolymers (three identical consecutive nucleotide bases or more), of which 14 thousand (3.3% of the total) disagreed on length between the two assemblies, resulting in alternative amino acid sequences for about 7% of the total 72,709 gene sequences evaluated. illumina cpg 450k pyrosequencing validation methylation imprinted genes imprinting 3), which is in agreement with previous results [5], [11]. PLOS ONE 7(3): 10.1371/annotation/64ba358f-a483-46c2-b224-eaa5b9a33939. We also found that the systematic single-base errors associated with GGC-motifs in Illumina data reported recently [16] represented only a minor fraction of the non-homopolymer-associated errors (0.015% of the total bases analyzed, consistent with the frequency reported in the original study). Although our metagenomic analysis is based on a single community sample, we believe it is robust and informative. Some of our results (e.g., assembly N50 comparisons, Fig. fantom illumina protocol solexa Illumina does not appear to share these limitations but it has its own systematic base calling biases [13]. In order to account for possible biases introduced by uneven genus abundance and provide statistically robust estimates, we employed a Jackknifing resampling method. methylation dna illumina array peripheral signatures cells breast cancer bead Although low coverage contigs (e.g., 1 to 5) are likely to contain a higher fraction of chimeric sequences than 0.2% according to our previous study [18], such contigs were rare in the results reported here, which included only contigs longer than 500 bp with average coverage 10 or higher (only about 3% of the contigs showed less than 5 coverage; Fig. Therefore, the two platforms provided comparable in situ abundances for the same genes or genomes. KyrpidesN, As evidence of this, analysis of the assemblies of isolate genomes that were sequenced using both platforms (see below) revealed that the extent of chimeric contigs, i.e., contigs that contained contaminating or in vitro generated sequences, in the Illumina and Roche 454 assemblies was, on average, less than 0.2% of the total length of the assembled contigs. sequencing wikipedi nanopore illumina We compared the two most frequently used platforms, the Roche 454 FLX Titanium and the Illumina Genome Analyzer (GA) II, on the same DNA sample obtained from a complex freshwater planktonic community. Base call errors and gap opening errors were identified as discrepancies between the read sequence and the reference assembly sequence using a custom Perl script. illumina We obtained (after trimming) a total of 502 Mbp (450 bp long reads) and 2,460 Mbp (100 bp pair-ended reads) from Roche 454 and Illumina sequencing, respectively, of the same community DNA sample. Finally, in all genomes analyzed, Illumina assemblies consistently recovered a larger percentage of the reference genome than Roche 454 assemblies (two tailed Whitney-Mann U test p-value=0.014; Fig. sequencing illumina synthesis implemented sequencers ligation probes labeled The sample comprised DNA from the prokaryotic fraction of a planktonic microbial community of a temperate freshwater lake (Lake Lanier, Atlanta, GA); the complexity of the community sampled (in terms of species richness and evenness) was estimated to be comparable to that of surface oceanic communities, but lower than that of soil communities [17]. Lanier.454 and Lanier.Illumina reads were trimmed at both the 5 and 3 ends using a Phred quality score cutoff of 20. School of Civil and Environmental Engineering, Georgia Institute of Technology, Atlanta, Georgia, United States of America, Affiliation View Next-generation sequencing (NGS) is commonly used in metagenomic studies of complex microbial communities but whether or not different NGS platforms recover the same diversity from a sample and their assembled sequences are of comparable quality remain unclear. We extracted the predicted gene sequences from the reads and the corresponding amino acid sequences were searched against the genes of the reference assembly of the same dataset using BLAT [28]. Among these genes, Roche 454 data appeared to have the wrong (artificial) sequence more often than Illumina data. Therefore, a desirable, first step in the analysis of metagenomic data frequently is to assemble sequences into longer contigs and, ultimately, into complete genome sequences.
Assemblies of isolate genome sequences (closed or high-draft) were downloaded from the NCBI RefSeq database (called reference assemblies for convenience); raw Illumina and Roche 454 sequencing reads were available through the Joint Genome Institute (JGI, www.jgi.doe.gov). No, Is the Subject Area "Gene sequencing" applicable to this article? 2). Conceived and designed the experiments: CL NK KTK. 3), low G+C% genomes sequenced with this platform may have 20% or more genes with frameshift errors whereas the Illumina platform is not affected as much by the G+C% of the sequenced DNA (Fig. It is possible that the remaining 10% of the contig sequences might have been different because of imperfect or uneven splitting of the original DNA sample into the two aliquots sequenced and the fact that the diversity in the sample was not saturated by sequencing (estimates based on rarefaction curves using raw reads indicated that we sampled about 8085% of the total diversity in the Illumina data). Thus, Roche 454 is advantageous with respect to gene calling when working with unassembled reads. Consistent with the metagenomic observations, we found that Roche 454 assemblies from genome data contained a significantly higher portion of frameshift errors compared to Illumina assemblies from the same genome, when the assemblies were built with 5 times more Illumina data than the Roche 454 data, matching the relative ratio of the metagenomic data reported above. We identified 0.4 million homopolymers (three identical consecutive nucleotide bases or more), of which 14 thousand (3.3% of the total) disagreed on length between the two assemblies, resulting in alternative amino acid sequences for about 7% of the total 72,709 gene sequences evaluated. illumina cpg 450k pyrosequencing validation methylation imprinted genes imprinting 3), which is in agreement with previous results [5], [11]. PLOS ONE 7(3): 10.1371/annotation/64ba358f-a483-46c2-b224-eaa5b9a33939. We also found that the systematic single-base errors associated with GGC-motifs in Illumina data reported recently [16] represented only a minor fraction of the non-homopolymer-associated errors (0.015% of the total bases analyzed, consistent with the frequency reported in the original study). Although our metagenomic analysis is based on a single community sample, we believe it is robust and informative. Some of our results (e.g., assembly N50 comparisons, Fig. fantom illumina protocol solexa Illumina does not appear to share these limitations but it has its own systematic base calling biases [13]. In order to account for possible biases introduced by uneven genus abundance and provide statistically robust estimates, we employed a Jackknifing resampling method. methylation dna illumina array peripheral signatures cells breast cancer bead Although low coverage contigs (e.g., 1 to 5) are likely to contain a higher fraction of chimeric sequences than 0.2% according to our previous study [18], such contigs were rare in the results reported here, which included only contigs longer than 500 bp with average coverage 10 or higher (only about 3% of the contigs showed less than 5 coverage; Fig. Therefore, the two platforms provided comparable in situ abundances for the same genes or genomes. KyrpidesN, As evidence of this, analysis of the assemblies of isolate genomes that were sequenced using both platforms (see below) revealed that the extent of chimeric contigs, i.e., contigs that contained contaminating or in vitro generated sequences, in the Illumina and Roche 454 assemblies was, on average, less than 0.2% of the total length of the assembled contigs. sequencing wikipedi nanopore illumina We compared the two most frequently used platforms, the Roche 454 FLX Titanium and the Illumina Genome Analyzer (GA) II, on the same DNA sample obtained from a complex freshwater planktonic community. Base call errors and gap opening errors were identified as discrepancies between the read sequence and the reference assembly sequence using a custom Perl script. illumina We obtained (after trimming) a total of 502 Mbp (450 bp long reads) and 2,460 Mbp (100 bp pair-ended reads) from Roche 454 and Illumina sequencing, respectively, of the same community DNA sample. Finally, in all genomes analyzed, Illumina assemblies consistently recovered a larger percentage of the reference genome than Roche 454 assemblies (two tailed Whitney-Mann U test p-value=0.014; Fig. sequencing illumina synthesis implemented sequencers ligation probes labeled The sample comprised DNA from the prokaryotic fraction of a planktonic microbial community of a temperate freshwater lake (Lake Lanier, Atlanta, GA); the complexity of the community sampled (in terms of species richness and evenness) was estimated to be comparable to that of surface oceanic communities, but lower than that of soil communities [17]. Lanier.454 and Lanier.Illumina reads were trimmed at both the 5 and 3 ends using a Phred quality score cutoff of 20. School of Civil and Environmental Engineering, Georgia Institute of Technology, Atlanta, Georgia, United States of America, Affiliation View Next-generation sequencing (NGS) is commonly used in metagenomic studies of complex microbial communities but whether or not different NGS platforms recover the same diversity from a sample and their assembled sequences are of comparable quality remain unclear. We extracted the predicted gene sequences from the reads and the corresponding amino acid sequences were searched against the genes of the reference assembly of the same dataset using BLAT [28]. Among these genes, Roche 454 data appeared to have the wrong (artificial) sequence more often than Illumina data. Therefore, a desirable, first step in the analysis of metagenomic data frequently is to assemble sequences into longer contigs and, ultimately, into complete genome sequences. 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abundance was determined based on the number and coverage of the contigs, as described elsewhere [17].  Protein-coding genes encoded in the assembled contigs were identified by the MetaGene pipeline [26]. LuoC, Further, the single-base sequence and gap opening error rates of individual reads were typically higher by 0.5% and a factor of 10, respectively, for the Roche 454 compared to the Illumina reads (Fig. More importantly, it is currently unclear how the above limitations affect the quality of the gene and genome sequences assembled from complex DNA samples, and whether the technologies provide different estimates of the genetic diversity in a sample due to their inherent chemistry and protocol differences. It should be noted, however, that most of the previous error estimates and sequencing biases have been determined based on relatively simple DNA samples (e.g., a single viral genome) and thus, their relevance for complex community DNA samples remains to be evaluated. Specifically, in genomes of about 50% G+C content (similar to the 47% G+C of the Lake Lanier metagenome), Roche 454 assemblies showed about 5% more frameshift errors than those of Illumina assemblies. An in-house package written in Python and Perl identified disagreements between Illumina and the reference Roche 454 reads associated with GGC motifs using the rules described previously [29] and counted the number of errors (scripts available upon request).
Protein-coding genes encoded in the assembled contigs were identified by the MetaGene pipeline [26]. LuoC, Further, the single-base sequence and gap opening error rates of individual reads were typically higher by 0.5% and a factor of 10, respectively, for the Roche 454 compared to the Illumina reads (Fig. More importantly, it is currently unclear how the above limitations affect the quality of the gene and genome sequences assembled from complex DNA samples, and whether the technologies provide different estimates of the genetic diversity in a sample due to their inherent chemistry and protocol differences. It should be noted, however, that most of the previous error estimates and sequencing biases have been determined based on relatively simple DNA samples (e.g., a single viral genome) and thus, their relevance for complex community DNA samples remains to be evaluated. Specifically, in genomes of about 50% G+C content (similar to the 47% G+C of the Lake Lanier metagenome), Roche 454 assemblies showed about 5% more frameshift errors than those of Illumina assemblies. An in-house package written in Python and Perl identified disagreements between Illumina and the reference Roche 454 reads associated with GGC motifs using the rules described previously [29] and counted the number of errors (scripts available upon request).
Affiliation
1C).  The matching gene of the assembly from the protein search using BLAT was compared to the gene matched by the raw read using Bowtie and instances of agreements (matched genes), disagreements (mismatched genes) and no match found (BLAT search did not match a gene while Bowtie mapping did) were counted and reported in Fig.
The matching gene of the assembly from the protein search using BLAT was compared to the gene matched by the raw read using Bowtie and instances of agreements (matched genes), disagreements (mismatched genes) and no match found (BLAT search did not match a gene while Bowtie mapping did) were counted and reported in Fig.  Samples were collected from Lake Lanier, Atlanta, GA, below the Browns Bridge in August 2009 and community DNA was extracted as described previously [17].
Samples were collected from Lake Lanier, Atlanta, GA, below the Browns Bridge in August 2009 and community DNA was extracted as described previously [17].  (D) Number of Roche 454 (x-axis) and Illumina (y-axis) reads mapping on the same contig shared between the two assemblies.
(D) Number of Roche 454 (x-axis) and Illumina (y-axis) reads mapping on the same contig shared between the two assemblies.  Department of Energy (DOE) Joint Genome Institute, Walnut Creek, California, United States of America, Affiliation Performed the experiments: CL DT. Similarly, the reference assembly sequence was cut into 500 bp long fragments and mapped onto assembled contigs longer than 500 bp; the unmapped regions of these contigs were identified as chimeric sequences and their total length (as a fraction of the total length of the contigs) represented the degree of chimerism for each dataset. Click through the PLOS taxonomy to find articles in your field. Roche 454 sequencing quality is evaluated in panels A through D, which show: (A) base call error rate of individual reads (x-axis) for each genome evaluated (y-axis); (B) base call error rate (y-axis) plotted against the G+C% of the genome; (C) gap opening error rate of individual reads (x-axis) for each genome evaluated (y-axis); (D) gap opening error rate (y-axis) plotted against the G+C% of the genome. 4), despite the fact that reads were trimmed based on the same quality standard prior to the analysis.
Department of Energy (DOE) Joint Genome Institute, Walnut Creek, California, United States of America, Affiliation Performed the experiments: CL DT. Similarly, the reference assembly sequence was cut into 500 bp long fragments and mapped onto assembled contigs longer than 500 bp; the unmapped regions of these contigs were identified as chimeric sequences and their total length (as a fraction of the total length of the contigs) represented the degree of chimerism for each dataset. Click through the PLOS taxonomy to find articles in your field. Roche 454 sequencing quality is evaluated in panels A through D, which show: (A) base call error rate of individual reads (x-axis) for each genome evaluated (y-axis); (B) base call error rate (y-axis) plotted against the G+C% of the genome; (C) gap opening error rate of individual reads (x-axis) for each genome evaluated (y-axis); (D) gap opening error rate (y-axis) plotted against the G+C% of the genome. 4), despite the fact that reads were trimmed based on the same quality standard prior to the analysis.